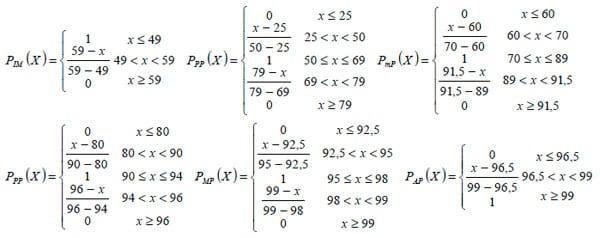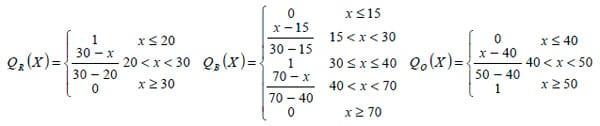1. INTRODUÇÃO
Uma das variações e particularidades do genoma da maioria das espécies são os chamados polimorfismos de base única (single nucleotide polymorphisms – SNPs), modificações de um único nucleotídeo, em uma dada sequência, quando comparada a outra. Ou seja, SNPs são pares de bases em uma única posição do DNA genômico que se apresentam com diferentes alternativas nas sequências (Figura 1) – isto é, alelos – e podem ser encontrados no genoma de indivíduos normais em algumas populações ou grupos de indivíduos.
A maior parte do genoma dos indivíduos de uma mesma espécie é idêntica, porém, existe a variabilidade genética, que consiste na alteração nas sequências de bases ao longo do DNA.
Tais alterações ocorrem por substituição, ausência ou duplicação de bases e os SNPs são o tipo mais comum de variabilidade genética (HapMap, 2003).
Figura 1: Exemplos hipotéticos de SNPs bi, tri e tetra-alélicos, respectivamente. A primeira linha, em negrito, representa a sequência consenso e as bases sublinhadas, os polimorfismos.
O que difere um indivíduo dos demais da sua espécie é o código genético, isto é, as sequências de nucleotídeos que formam as moléculas e sequências de DNA, RNA e proteínas, que, por sua vez, interagem e formam as células, as quais, por sua vez, formam os tecidos, os órgãos, até que, finalmente, formam os indivíduos. Ou seja, as diferenças se iniciam na ordem em que os nucleotídeos se apresentam e essa é a importância dos SNPs, já que a alteração de um único nucleotídeo em uma dada sequência pode alterar a produção de uma certa proteína. Assim, eles são importantes no estudo da variabilidade das espécies, uma vez que podem provocar alterações funcionais ou fenotípicas, que podem implicar em consequências evolutivas ou bioquímicas nos indivíduos em que os SNPs se manifestam.
2. OBJETIVO
O objetivo deste trabalho é o de apresentar e discutir um modelo de inferência difusa para suporte à decisão na descoberta de SNPs em sequências de cDNA, permitindo sua implementação por meio de um modelo computacional, fundamentado em aprendizado de máquina para descoberta de conhecimento em bases de dados (knowledge discovery in database – KDD).
3. MATERIAIS E MÉTODOS
A discussão do modelo proposto passa pela obtenção e montagem de sequências de cDNA, a busca de SNPs nessas sequências e, ainda, a modelagem computacional para suporte à decisão por meio de inferência difusa, as quais são abordadas na presente seção.
A partir da montagem de sequências-consenso, pelo alinhamento de sequências obtidas por algum procedimento de análise e interpretação do DNA genômico, se inicia a investigação em sequências de cDNA. Neste trabalho, o procedimento utilizado foi a geração de sequências expressas identificadas (expressed sequence tags – EST), também conhecidas como sequências derivadas de transcritos, que são medições dos níveis de mRNA (Lesk, 2005), isto é, a porção extraída do RNA transcrito e traduzida em proteína, a qual gera sequências curtas de cDNA. O sequenciamento de ESTs baseia-se em "capturar" essas sequências de nucleotídeos de regiões expressas do genoma que correspondem sequências com, aproximadamente, 200 e 500 pares de bases (Adams et al., 1991), o que torna necessário agrupar (cluster) e alinhar as sequências obtidas que correspondem a fragmentos de um mesmo gene, para que seja possível montar sequências mais extensas.
A descoberta de SNPs por algoritmos computacionais é uma prática bastante difundida e os programas Polyphred e Polybayes se destacam pelo seu amplo uso nessa área. O Polyphred analisa os sinais expressos no sequenciamento do material genético e detecta SNPs a partir da variação dos sinais de fluorescência dos cromatogramas, procurando por reduções nas regiões do pico do sinal onde uma segunda base foi detectada (Nickerson et al., 1997). Por sua vez, o Polybayes analisa as bases geradas a partir da "leitura" dos cromatogramas – que nomeia e atribui um valor de qualidade (Phred quality score – PQS) para cada base (Ewing et al., 1998) – e utiliza um algoritmo de inferência Bayesiana, que procura por seções transversais onde as sequências alinhadas apresentam bases diferentes entre si (Marth et al., 1999).
Os referidos programas implementam diferentes avaliações sobre diferentes atributos. Contudo, espera-se que apresentem resultados similares, ao tratarem um mesmo conjunto de sequências, mas, não é incomum fornecerem resultados diferentes, o que produz incerteza na tomada de decisão. Além disso, deve ser notado que esses dois programas têm seus resultados influenciados pelo PQS, obtido durante a leitura dos cromatogramas.
Modelos de inferência difusa são adequados para representar a informação imprecisa, a qual pode ser expressa por um conjunto de regras lingüísticas. Caso exista a possibilidade de que os operadores sejam organizados como um conjunto de regras da forma
se ANTECEDENTE então CONSEQUENTE,
o raciocínio subjetivo pode ser construído em um algoritmo computacionalmente executável (Tanscheit, 2007), com capacidade de classificar, de modo impreciso, as variáveis que participam dos termos antecedentes e consequentes das regras em conceitos qualitativos, e não quantitativos, o que representa a idéia de variável linguística (Almeida et al., 2005).
Trabalhar com valores incertos possibilita a modelagem de sistemas complexos, mesmo que se reduza a precisão do resultado, o que não retira a credibilidade. Se as incertezas, quando consideras isoladamente, são indesejáveis, quando associadas a outras características dos sistemas a serem modelados, em geral, permitem a redução da complexidade do sistema e aumentam a credibilidade dos resultados obtidos (Klir et al., 1995).
A subjetividade no raciocínio, sendo transmitida e compreendida entre interlocutores, é expressa em "termos e variáveis linguísticas" (Zadeh, 1973), mas não é expressa pela lógica clássica ou qualquer abordagem matemática tradicional. Por exemplo, o uso de adjetivos que representam imprecisão ou incerteza, tais como alto e baixo, ou, ainda, relações, como o conjunto das pessoas altas, não podem ser expressos por essas abordagens, a menos que seja definido, por exemplo, a altura a partir da qual uma pessoa pode ser considerada alta.
4. MODELO PROPOSTO
Cada ponto possivelmente polimórfico identificado pelo Polyphred ou pelo Polybayes tem a sua probabilidade estimada por cada um dos programas e o seu valor da qualidade da base – Phrap quality (PQ) – na sequência-consenso, que não é considerada diretamente por esses aplicativos. Com os resultados do Polyphred e do Polybayes, o modelo de inferência auxilia a tomada de decisão, no caso em que as informações sejam divergentes e na confirmação de casos coincidentes, avaliando esses resultados. O modelo ainda inclui o PQ como um "valorizador" adicional, que reduz os efeitos específicos de cada um dos programas e amplia as possibilidades de investigação. PQ é utilizado na análise como apoio à decisão, então, aos dados prévios sobre a possibilidade de o ponto vir a ser um SNP, acrescenta-se a sua qualidade, com o objetivo de se estabelecer uma das três possibilidades: a confirmação de o ponto ser um SNP (SNP confirmado – SNPC), a eliminação dessa possibilidade (SNP descartado – SNPD) ou uma situação sem a confirmação dessa possibilidade, mas também sem elementos conclusivos para seu descarte (SNP não confirmado – SNPNC). No modelo proposto, as funções de pertinência adotadas foram baseadas:
1. No Polyphred score (PPS5), que estabelece seis classes com intervalos precisos (Nickerson et al., 1997), variando de 1, que indica um PPS >= 99 e uma taxa de verdadeiros positivos de 97%, sendo provável a existência de SNPs; até 6, que indica PPS <= 49 e uma taxa de verdadeiros positivos de 1%, sendo improvável a existência de SNPs. Assim, a sua função de pertinência foi definida pela variável linguística probabilidade, com os termos: improvável (PIM), pouco provável (PPP), medianamente provável (PmP), provável (PPR), muito provável (PMP) e altamente provável (PAP);
2. No PQ, que varia entre 4 e 90 e é separado, pelo limiar6 PQ = 20, em duas classes de valores; sendo sua função de pertinência definida como a variável linguística qualidade,
nos termos: ruim (QR), boa (QB) e ótima (QO).
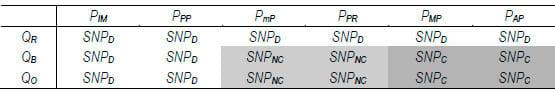
Essas variáveis são utilizadas em trinta e seis regras de inferência, divididas em dois conjuntos, que possuem a respectiva metade para avaliar qualidade e probabilidade, segundo Polyphred e Polybayes, e que relacionam as combinações possíveis dos termos linguísticos:
Cada um desses dois conjuntos inferem as decisões representadas na Tabela 1.
Tabela 1: Decisões inferidas pelo modelo a partir da avaliação de qualidade e probabilidade.
5. RESULTADOS E DISCUSSÃO
Os termos e variáveis linguísticas aumentam a complexidade de um sistema de computação frente à capacidade de trabalharem com números ou outros valores exatos, discretos, muitas vezes, excludentes e o modelo proposto, definido a partir de técnicas de aprendizado de máquina, substitui, por meio de inferência difusa, medidas de probabilidade, determinadas pelo Polyphred e Polybayes, contínuas no intervalo [0, 1] e associadas à possibilidade de um ponto vir a ser um SNP, por um outro atributo, que permite decidir sobre a classe de cada ponto – SNPC, SNPD ou SNPNC – e essa decisão pode ser tomada devido à possibilidade de o modelo de inferência difusa refletir o raciocínio subjetivo a partir de regras de inferência.
O modelo proposto, além da fundamentação apresentada, foi determinado pelas características dos resultados obtidos com ferramentas usuais de identificação de SNPs em que critérios fixos e precisos – como o PPS – não são adequados, quando são obtidos resultados próximos à divisão de classes ou quando essas ferramentas apresentam resultados conflitantes.
Uma ampla discussão sobre o modelo proposto pode ser encontrada em Arbex (2009), juntamente com a descrição da ferramenta fuzzyMorphic.pl, utilizada para o desenvolvimento e a implementação desse modelo.
6. CONCLUSÃO
Fundamentado na lógica difusa, foi elaborado um modelo de inferência para suporte à decisão, pela análise de resultados prévios e frente a características de incerteza, que são comuns em processos decisórios.
7. REFERÊNCIAS
ADAMS, M. D.; KELLEY, J. M.; GOCAYNE, J. D.; et al. Complementary DNA sequencing: expressed sequence tags and human genome project, Science, v. 252, n. 5013, p. 1651-1656, jun. 1991.
ALMEIDA, P. E. M.; EVSUKOFF, A. G. Sistemas fuzzy. In: REZENDE, S. O. (ed.). Sistemas inteligentes: fundamentos e aplicações. Barueri: Manole, 2005. p. 169–202.
ARBEX, W. Modelos computacionais para identificação de informação genômica associada à resistência ao carrapato bovino, 2009. Tese de doutorado. Universidade Federal do Rio de Janeiro. 200 p.
EWING, B.; HILLIER, L.; WENDL, M. C.; et al. Base-calling of automated sequencer traces using Phred (I): Accuracy assessment, Genome Research, v. 8, p. 175-185, 1998.
HAPMAP The International HapMap Project, Nature, v. 426, n. 6968, p. 789-796, dec. 2003. (The International HapMap Consortium)
KLIR, G. J.; YUAN, B. Fuzzy sets and fuzzy logic: theory and applications. Upper Saddle River: Prentice Hall, 1995. 592 p.
LESK, A. M. Introduction to bioinformatics. New York: Oxforf University Press, 2nd ed, 2005. 378 p.
MARTH, G. T.; KORF, I.; YANDELL, M. D.; et al. A general approach to single-nucleotide polymorphism discovery, Nature Genetics, v. 23, p. 452-456, dec. 1999.
NICKERSON, D. A.; TOBE, V. O.; TAYLOR, S. L. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing, Nucl. Acids Res., v. 25, n. 14, p. 2745-2751, 1997.
TANSCHEIT, R. Sistemas fuzzy. In: OLIVEIRA JR., H. A. (ed.). Inteligência computacional: aplicada à administração, economia e engenharia em Matlab. São Paulo: Thomson Learning, 2007, pp. 229-264.
ZADEH, L. A. Outline of a new approach to the analysis of complex systems and decision processes, IEEE Trans. on Systems, Man, and Cybernetics, v. SMC-3, p. 28-44, 1973.