Acetonitrila Laboratórios Cromatografia
Metodologia para Tratamento de Acetonitrila Comercial para uso em Laboratórios de Cromatografia Líquida.
Publicado: 19 de novembro de 2012
Por: João Oiano Neto; Humberto Ribeiro Bizzo; Ronoel Luiz de Oliveira Godoy; Marco Antonio Silva Pinto; Jeane Santos da Rosa; Manuela Cristina Pessanha de Araujo; Sidney Pacheco.
Introdução.
A acetonitrila (CH3CN; M.M. 41,05 g/mol; bp1atm 81,6°C) é produzida industrialmente pela desidratação da acetamida ou pela reação entre acetileno e amônia (WINDHOLZ et al., 1983). Representa um subproduto na produção da acrilonitrila, monômero de síntese da poliacrilonitrila, que é utilizada na fabricação de fibras têxteis acrílicas. É utilizada quando se necessita de um solvente orgânico com grande constante dielétrica, em meios reacionais que requerem ionização, ou como solvente em titulações não aquosas (WINDHOLZ et al., 1983).
A acetonitrila é um solvente orgânico de ampla aplicação em laboratórios analíticos que utilizam a técnica de cromatografia líquida de alta eficiência (CLAE), com colunas cromatográficas de fase reversa aminopropil sílica (NH2), octilsilano (C8), octadecilsilano (C18), etc.
Nas análises de açúcares (sacarose, glicose, frutose) é o solvente de escolha com colunas aminopropil sílica (NH2) e detector de índice de refração (RI), pois, misturada à água em diferentes proporções, resulta em boa seletividade cromatográfica (NOLLET, 1992).
Outra vantagem do uso da acetonitrila como solvente em análises cromatográficas por CLAE reside no fato de ser "transparente", quando se utilizam detectores que operam na região do ultravioleta-visível (UV-vis.).
Possui um valor de cut-off (absortividade máxima) menor que 0,01 a 230nm, sendo transparente em comprimentos de onda acima desse valor. Sua absortividade máxima, correspondendo ao cromóforo nitrila, ocorre a 160nm (SNYDER; KIRKLAND; GLAJCH, 1997).
No auge da atual crise econômica do mercado financeiro, a acetonitrila grau HPLC, vendida a R$ 250,00 até este momento, apresentou uma alta no seu valor e passou a ser comercializada a R$ 2.650,00; representando um aumento de preço de aproximadamente 960%.
Os processos de purificação da acetonitrila comercial podem envolver várias etapas, com sucessivas destilações sob pentóxido de fósforo - P2O5 para eliminação do azeótropo formado com a água (MORITA; ASSUMPÇÃO, 2001) ou agitação com hidróxido de sódio aquoso, seguido de sucessivas lavagens com água, adição de sulfato de sódio anidro e destilação sob pentóxido de fósforo, conforme descrito por Perrin e Armarego (1988).
Desta forma, o objetivo deste trabalho foi desenvolver uma metodologia alternativa para o tratamento da acetonitrila, de forma rápida e barata, sem envolver uma série de etapas e reagentes.
Experimental
Amostras
Os frascos de acetonitrila, com prazo de validade vencido há vários anos, encontravam-se armazenados no almoxarifado da Embrapa Agroindústria de Alimentos e foram divididos em três lotes: LOTE 1- acetonitrila grau HPLC marca A; LOTE 2 - acetonitrila grau analítico marca A e LOTE 3 - acetonitrila grau analítico marca B.
Filtração em Alumina
Nesta etapa, seis litros de acetonitrila foram filtrados à pressão reduzida em um funil de vidro (d.i. 11cm) com placa sinterizada, contendo um leito de aproximadamente 7,0cm de espessura de alumina neutra Merck® 90 (63-200 mm) ativada a 120oC em estufa por 12 horas (Figura1).
Neste processo, utiliza-se uma camada de lã de vidro para evitar que a aplicação do solvente, diretamente sobre o leito de alumina, danifique seu empacotamento.
Figura 1. Sistema para filtração à pressão reduzida. (1) lã de vidro; (2) leito de alumina neutra; (3) funil de vidro com placa sinterizada; (4) adaptador de borracha; (5) garra com suporte universal; (6) kitassato e (7) mangueira de vácuo.

A etapa de filtração foi efetuada duas vezes para cada lote, a fim de remover possíveis contaminantes mais polares que a acetonitrila, como compostos fenólicos, segundo o descrito por Perrin e Armarego (1988).
Após o processo de filtração, a alumina utilizada foi recuperada (para posterior reutilização) mediante lavagem com, aproximadamente, 300mL de metanol grau HPLC, seguidos de 500mL de água ultrapura.
A lavagem com metanol foi feita até não se observar mais a presença da coloração amarela no metanol eluído do funil. Em seguida, a alumina foi ativada a 120°C em estufa por 12 horas.
Destilação
A destilação do solvente, obtido após a filtração em leito de alumina, foi realizada em um sistema de destilação fracionada semi automatizado modelo NGW, com controle digital da temperatura de ebulição (Figura 2).
Figura 2. Sistema para destilação fracionada. (1) coluna de destilação; (2) manta de aquecimento; (3) balão de fundo redondo; (4) controlador de temperatura; (5) controlador de pressão; (6) banho termostatizado para água de refrigeração

Análise por CG-EM
A análise da acetonitrila foi feita após as etapas de filtração em alumina e destilação fracionada, utilizandose um cromatógrafo a gás modelo Agilent® 6890 equipado com um espectrômetro de massas Agilent® 5973N, com fonte de impacto eletrônico (EI) a 70eV.
Na análise cromatográfica, utilizou-se uma coluna HP5 (5% fenil + 95% dimetilsilicone), L 30m, d.i 0.25mm e 0.25mm de espessura de filme, volume injetado de 1mL em splitless, Tinj. 250°C, Tini. 40°C, tini. 5min., gradiente de temperatura 40-150°C/5°C por min., 150-240°C/10°C por min. e hélio como gás de arraste a 1,0mL/min.
A confirmação da identidade química dos componentes das amostras foi feita pela comparação de relação massa/carga (m/z) dos íons com os dados da biblioteca do equipamento.
Para medida do percentual relativo dos compostos presentes na acetonitrila do LOTE 3, após a destilação, utilizou-se a mesma condição cromatográfica, porém com um detector FID (Flame Ionization Detector).
Resultados e Discussão
Análise por CG-EM dos Lotes 1 e 2
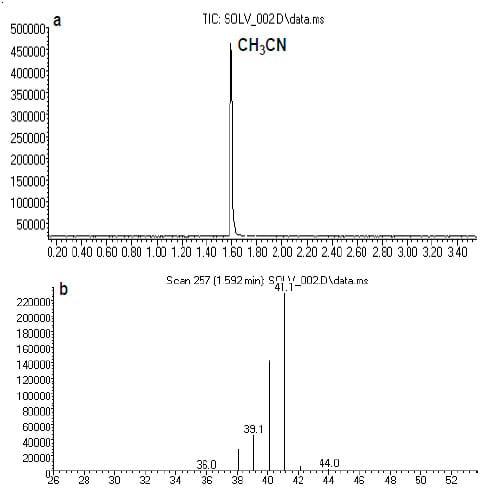
A análise por CG-EM da acetonitrila do LOTE 1, submetido apenas à filtração, apresentou apenas o pico relativo à acetonitrila (Figura 3).
Nesta análise, a acetonitrila apresentou o pico do íon molecular [M.+] com uma relação marga/carga (m/z) igual a 41.1.
Figura 3. Cromatograma da análise por CG-EM da acetonitrila LOTE 1 (a); espectro de massas da acetonitrila (b).
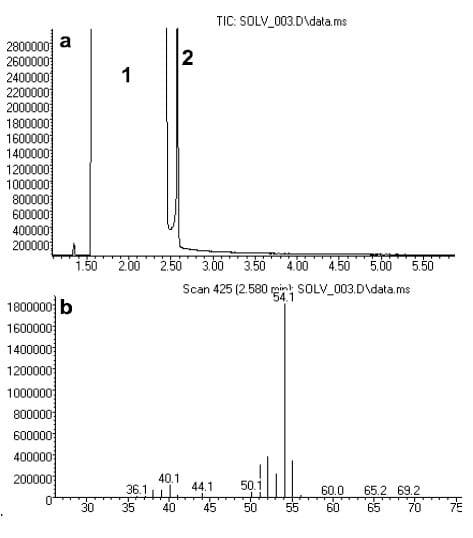
A análise da acetonitrila do LOTE 2, submetida apenas à filtração (Figura 4), revelou, além do pico relativo à acetonitrila, o pico indicativo da presença da propanonitrila (CH3CH2CN).
Nesta análise, a propanonitrila (M.M. 55g/mol) apresentou um fragmento com marga/carga (m/z) igual a 54.1, correspondente ao íon [M+-H].
Esses lotes foram testados no Laboratório de CLAE como fase móvel durante a análise de açúcares. Não se observou qualquer mudança no tempo de retenção dos açúcares de interesse (sacarose, glicose, frutose), na seletividade e resolução cromatográfica.
Mediante comparação desses testes com os obtidos em análises prévias, utilizando-se acetonitrila grau HPLC, verificou-se que a acetonitrila de ambos os lotes não causou qualquer alteração nos resultados analíticos.
Para estes lotes, não foram necessárias etapas posteriores de purificação após a filtração em alumina.
Figura 4. Cromatograma da análise por CG-EM da acetonitrila LOTE 2 (a) e espectro de massas da propanonitrila (b); onde (1) CH3CN e (2) CH3CH2CN.
O tratamento de ambos os lotes (~8L) representou um economia aproximada de R$ 5.300,00 com a compra deste solvente.
Análise Lote 3
Ao final da filtração, pode-se observar que a limpeza da alumina com metanol produziu uma solução de coloração amarelada intensa, indicando que este lote estava contaminado com outros compostos insaturados.
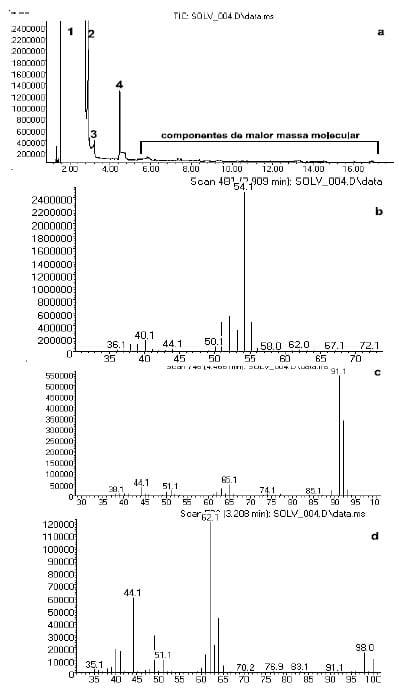
A análise por CG-EM de uma alíquota do solvente filtrado (Figura 5) mostrou a presença dos picos referentes à propanonitrila - CH3CH2CN (tR 2,9min.), ao 1,2- dicloroetano - Cl(CH2)2Cl (tR 3,2min.) e ao tolueno - C7H8 (tR 4,4min.), além de outros compostos de maior massa molecular não identificados.
A propanonitrila apresentou um fragmento com m/z igual a 54.1, correspondente ao íon [M+-H]. O tolueno (M.M 92g/mol) apresentou um fragmento com m/z igual a 91.1, referente à formação do íon tropílio [M+-H], ao passo que o 1,2-dicloroetano (M.M 98g/mol) apresentou dois fragmentos distintos, com relação marga/carga (m/z) igual a 98.0 e 62.1, referente à formação dos íons [M.+] e [M+-HCl], respectivamente.
Figura 5. Cromatograma da análise por CG-EM da acetonitrila LOTE 3 (a), espectro de massas da propanonitrila (b), (c) tolueno e (d) 1,2-dicloroetano; onde (1) CH3CN, (2) CH3CH2CN, (3) Cl(CH2)2Cl e (4) C7H8.
Em função destes resultados, este lote de solvente foi submetido à destilação fracionada, com posterior análise por CG-EM com detector FID.
A análise do destilado indicou, novamente, a presença de propanonitrila, 1,2-dicloroetano e tolueno em mistura com a acetonitrila. No entanto, pode-se verificar a completa remoção dos compostos de maior massa molecular, após a destilação fracionada. Em conjunto, estes contaminantes orgânicos representam uma proporção relativa na mistura de 0,06%.
A não remoção destes compostos, na etapa de destilação fracionada, pode ser justificada pela formação de um possível azeótropo com a água, devido ao caráter higroscópico da acetonitrila.
No entanto, o percentual relativo destes compostos é muito baixo (0,06%), equivalendo ao encontrado na acetonitrila comercial grau HPLC de 0,1%. Neste valor percentual não está inserido o teor relativo de água, uma vez que a mesma não é quantificada em um detector FID.
Este lote será testado, posteriormente, na análise de açúcares, com o intuito de observar se a presença de tais compostos interfere na eficiência da separação cromatográfica.
Caso isso seja observado, como alternativa adicional ao processo de purificação, este lote será submetido à destilação fracionada sob vácuo, para verificar se este procedimento evita a formação do azeótropo, com consequente eliminação desses contaminantes.
Conclusões
O método desenvolvido mostrou-se rápido e de fácil execução, permitindo o tratamento de grandes quantidades de acetonitrila em pouco tempo.
A acetonitrila obtida ao final do processo apresentou características apropriadas para ser utilizada em análises de açúcares por cromatografia líquida.
A recuperação e reutilização da alumina no processo de filtração contribuem para a diminuição dos custos desta metodologia.
Além de contribuir para a redução dos custos com a compra deste solvente em períodos de escassez e alta de preços, este procedimento gera uma significativa economia, evitando o envio de grandes quantias de solvente, fora do prazo de validade, para descarte e incineração.
Referências
MORITA, T.; ASSUMPÇÃO, R. M. V. Manual de soluções, reagentes e solventes: padronização, preparação, purificação. 1. ed. São Paulo: E. Blücher, 2001. 630 p.
NOLLET, L. M. L. (Ed.). Food analysis by HPLC. 1st ed. New York: M. Dekker, 1992. 759 p.
PERRIN, D. D.; ARMAREGO, W. L. F. Purification of laboratory chemicals. 3rd ed. Oxford; New York: Pergamon Press, 1988. 391 p.
SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Practical HPLC method development. 2nd ed. New York: J. Wiley, 1997. 765 p.
WINDHOLZ, M.; BUDAVARI, S.; BLUMETTI, R. F.; OTTERBEIN, E. S. (Ed). The Merck index : an encyclopedia of chemicals, drugs, and biologicals. 10th ed. Rahway, NJ: Merck, 1983.
Autores:

Embrapa

Embrapa
Embrapa
Embrapa
Mostrar mais
Recomendar
Comentário
Compartilhar

Quer comentar sobre outro tema? Crie uma nova publicação para dialogar com especialistas da comunidade.

